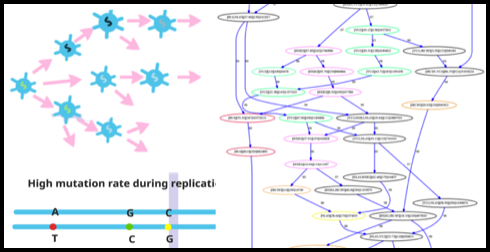
A patient with chronical infection of RNA viruses (such as HIV, HCV etc.) can generate billions of new viral particles each day. Of these particles, there are an unknown number of different genomes, which can act together to escape the host’s immunity system. For example, some of these genomes might contain genes with resistance to anti-viral drugs and thus are more likely to lead to treatment failure. Therefore, being able to characterize these different genomes and their abundance is essential for vaccine and anti-viral drug design.
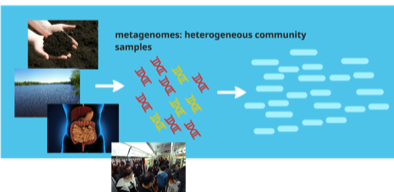
Metagenomic sequencing provides us new opportunities to study the viral populations of both new and known viruses. We are interested in characterizing different viral genomes inside viral populations using metagenomic data. One of the biggest challenges in this field is genome-scale haplotype reconstruction, which aims to reconstruct the whole genomes of each different strain. As the genomes inside the same viral population can be highly similar and have heterogeneous sequencing coverage, no many features are useful to distinguish genomes of strains. It is common that only gene-wise or local regions of genomes can be output by existing tools. Thus, we are working on developing better algorithms to assembly short reads into different haplotypes from viral metagenomic data.
We recently developed a new overlap graph-based de novo haplotype reconstruction tool PE-Haplo. https://github.com/chjiao/OL_PEHaplo.
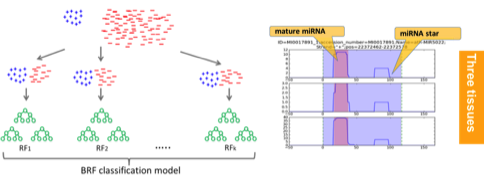
Metagenomic sequencing has clear advantages over conventional methods. It contains nucleic acid sequences of both culturable and unculturable microbial species and of both new and known species. We are particularly interested in identifying targeted genes in pathways that are of interest to users. Thus, we combine available reference genes/protein families with de novo sequence assembly. The output will be the detection of the genes and also their abundance in a sample. Our projects: SAT-assembler [Plos-computational biology, 2014], SALT [Bioinformatics, 2013], CRISPR [ECCB & Bioinformatics, 2016], Xander [collaborative work, Microbiome, 2015], FrameBot [collaborative work, mBio, 2013]. Genome annotation is to label segment of a genome sequence with its possible properties, structures, and functions. We are particularly interested in noncoding RNAs, such as microRNAs (miRNAs) and lncRNAs. We used a random forest model to distinguish protein-coding genes from lncRNAs by incorporating novel features. We also developed an efficient tool for fast miRNA and specific plant miRNA prediction using small RNA-Seq data. By carefully incorporating both sequence, structural, and expression-based features of miRNAs, our tool is more accurate in miRNA prediction than other benchmarked tools. In addition, it is both memory and CPU efficient and thus can be applied to large-scale RNA-seq data of large plant genomes. The miRNA prediction tool is freely available at: https://github.com/hangelwen/miR-PREFeR Gene prediction is a key step in genome annotation. Ab initio gene prediction enables gene annotation of new genomes regardless of availability of homologous sequences. There are a number of existing ab initio gene prediction tools and they have been widely used for gene annotation for various species. However, existing tools are not optimized to identify genes with highly variable GC content. In addition, many grass genes exhibit a sharp 5′-3′ decreasing GC content gradient, which is not carefully modeled by existing gene prediction tools. As a result, these tools have limited sensitivity and accuracy for predicting genes with GC gradients. Thus, there is a need for a tool that can effectively predict those genes with highly variable GC contents without manual intervention.
We designed and implemented a new hidden Markov model (HMM)-based ab initio gene prediction tool, which is optimized for finding genes with highly variable GC contents, such as the genes with negative GC gradients in grass genomes. We tested the tool on three datasets from Arabidopsis thaliana and Oryza sativa. The results showed that our tool can identify genes missed by existing tools due to the highly variable GC contents. GPRED-GC is best used for identifying genes with highly variable GC contents. It provides a useful supplementary tool to existing ones such as Augustus for more sensitive gene discovery. The source code is freely available at https://sourceforge.net/projects/gpred-gc/.